Python package for microsatellite genotyping from amplicon sequencing data

Project description



Python package for microsatellite genotyping from highly multiplexed amplicon sequencing data
Features
Semi-automatic genotyping optimized for amplicon sequencing data of microsatellite loci
Visual genotyping with interactive plots
Fast SSR search in sequences
Automatic grouping and naming of alleles based on polymorphisms in both SSR and non-SSR regions
Support for multi-core processing
Requirements
Installation
Install from PyPI
pip install massgenotypingInstall the latest version from a Git repository
pip install git+https://github.com/kohyamat/massgenotypingUsage
mgt [-h] SUBCOMMAND [OPTIONS]Subcommand list:
demultiplex: demultiplex raw amplicon sequences based on primer sequencesmerge-pairs: merge paired-end readsdenoise: reduce any noise that may have been generated during sequencing and PCRfilter: filtering for erroneous sequence variants and screening for putative allelesallele-check: check allele candidates and create an allele databaseallele-call: assign alleles to raw amplicon sequencesshow-alignment: show a sequence alingment
The details of the options for each subcommand can be checked by mgt SUBCOMMAND -h.
Tutorials with example data
Here’s a step-by-step tutorial using the example data.
1. Demultiplex raw amplicon sequences based on primer sequences
As a first step, the sequence data is split based on the primer sequence. The input can be one or two sequence files in the FASTQ format, or a directory containing multiple sequence files. Primer sequences can be read from CSV or FASTA files. Please check the example data for the format of the input data.
mgt demultiplex examples/sequence_data -g "*_R[12]_*" -m examples/marker_data.csvThe result files are written in subdirectories within the output directory (./project by default) for each marker.
2. Merge paired-end reads and trim primer sequecnes
For the paired-end sequencing data, the respective sequence pairs are merged using NGmerge program. The following command removes the the primer sequences after merging sequence pairs.
mgt merge-pairs ./project -m examples/marker_data.csv --trim-primerFor single-end data, this step can be skipped. The removal of the primer sequence can also be performed in the step 1.
3. Reduce noise (optional but recommended)
This step corrects any noise (very low-frequency point mutations) that may have been generated during sequencing or PCR. This step is not necessarily required, but it will make the following step easier.
mgt denoise ./project/*/*_merged.fastq.gz4. Filter out erroneous sequence variants
In this step, the sequence of putative alleles is extracted for each marker in each sample,
while removing any erroneous sequence variants, such as ‘stutter’ sequences.
After some rough filtering, an interactive plot allows you to choose which sequence variants to keep.
You can skip this visual-checking procedure with the --force-no-visual-check option.
mgt filter ./project -m examples/marker_data.csv5. Check a multiple sequence alignment and make an allele database
The database is created after checking the alignment of the putative allele sequences. If necessary, you can further filter out the erroneous sequence variants.
mgt allele-check ./project6. Assign alleles to raw amplicon sequences
Finally, the following command perform a BLASTn search against the database created for each marker and assign alleles to the raw sequence data. The genotype tables are created within the output directory.
mgt allele-call ./project -m examples/marker_data.csvScreenshots
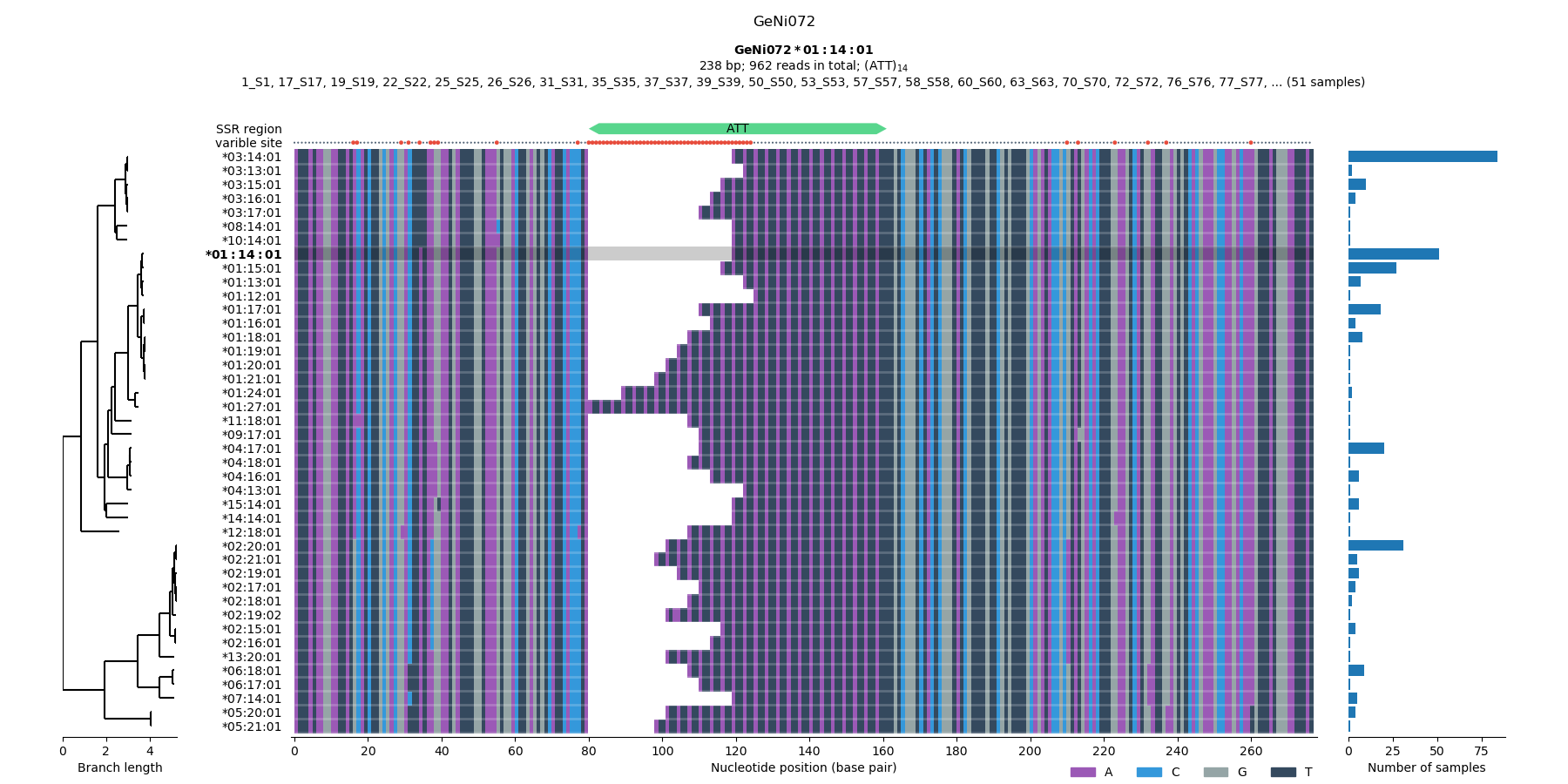
Figure 1. Checking the multiple sequence alignment across the samples (STEP 5).
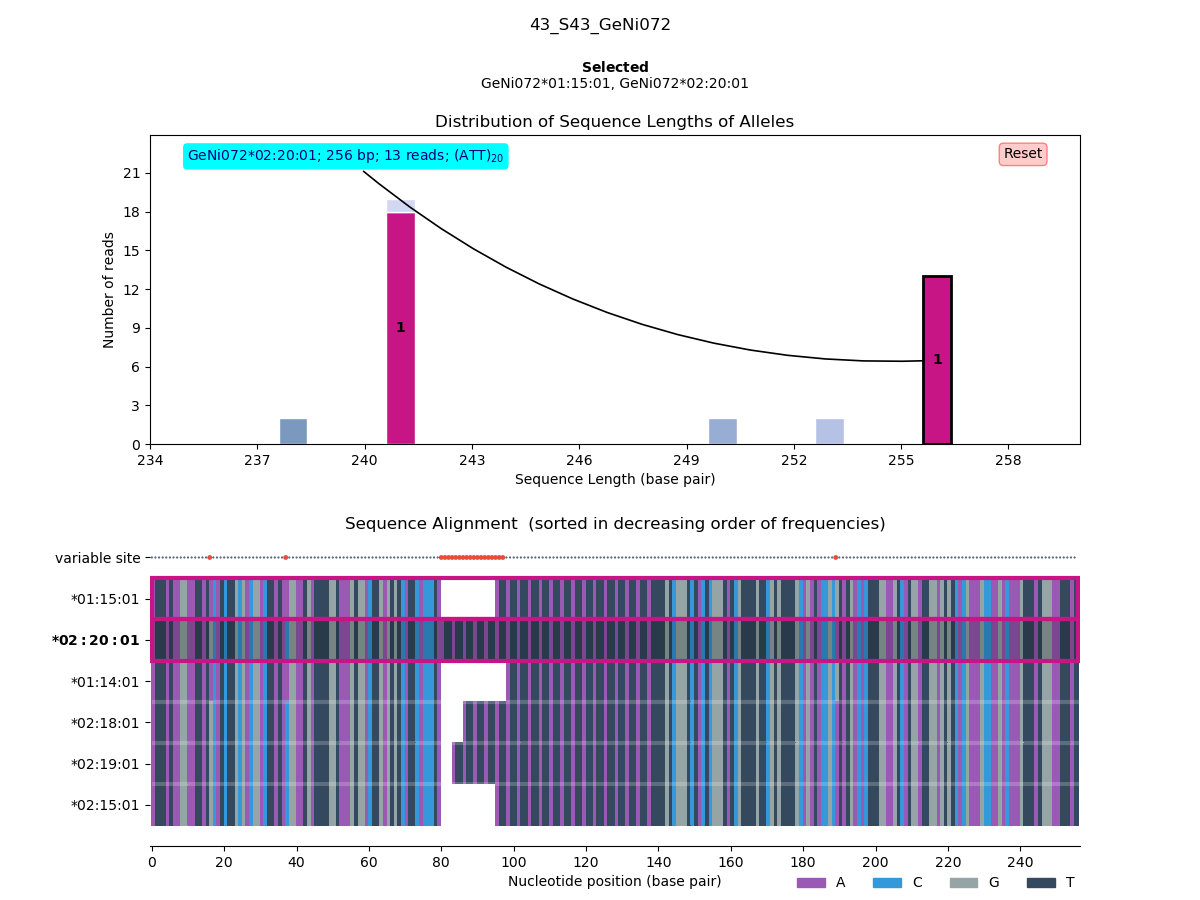
Figure 2. Visual genotyping (STEP 6).
Contributing to massgenotyping
Contributions of any kind are welcome!
License


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Hashes for massgenotyping-0.2.2-py3-none-any.whl
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 29a6b85f866970e75c4e03cee78d98af503d233ca3ab4ba9f0c230129926a0c5 |
|
| MD5 | affbb5ea340b14661bd07af75e4bcc9b |
|
| BLAKE2b-256 | fe05c9f7c84f394f5530b36fb7d628c9d2f15e9feac0433d8258c72d45ec72e5 |







